Technology.
de novo sequencing algorithm
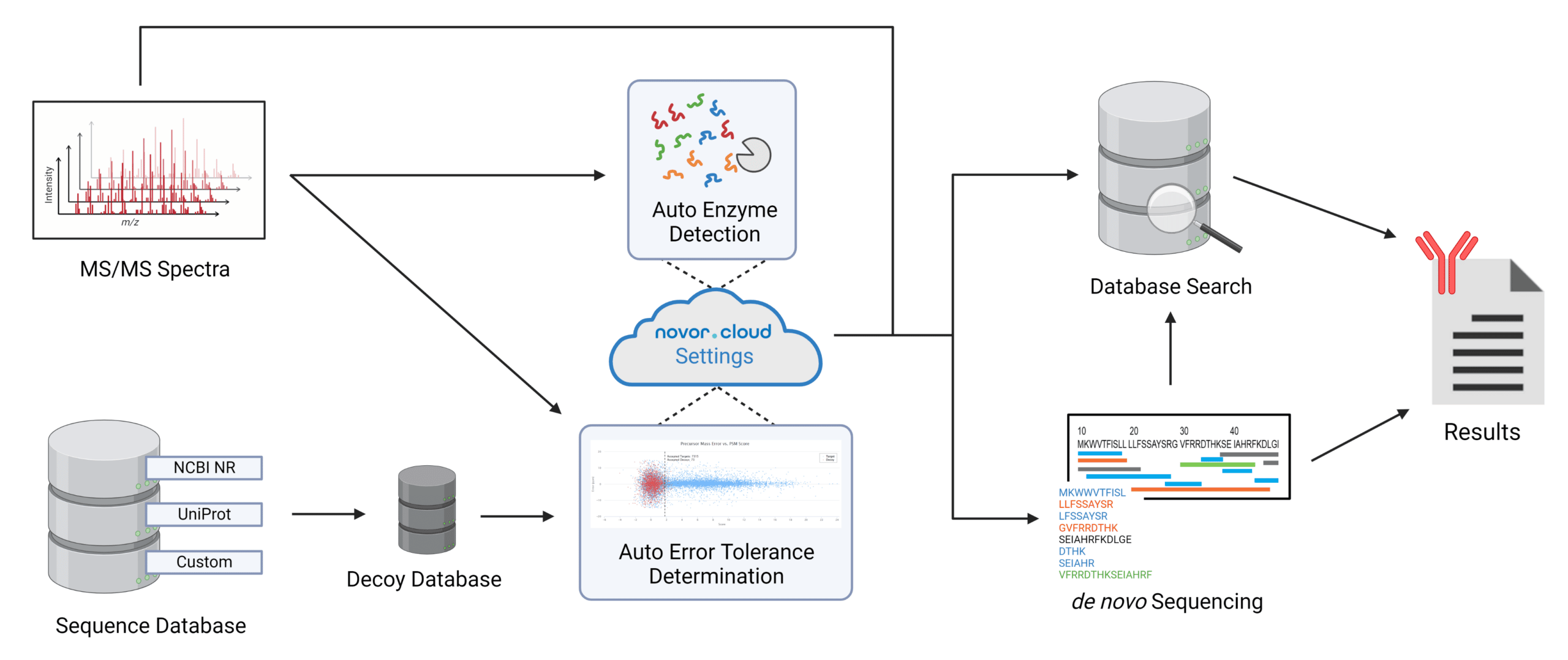
Novor.cloud launched in 2021, evolved from the original ‘novor’ de novo sequencing algorithm, written in 2015 by Prof. Bin Ma, who is now President and Chief Scientist at Rapid Novor. Novor.cloud is the culmination of 20+ years of de novo sequencing, protein identification, mass spectrometry and bioinformatics R&D. Prof. Ma is also the author of the older PEAKS de novo sequencing algorithm in 20031,2, which is still the most widely used de novo sequencing software today and one of the most popular database search tools. Unlike earlier de novo sequencing algorithms, which rely on heuristics and aren’t fast, the novor.cloud de novo sequencing algorithm uses artificial intelligence and decision trees to optimize scoring and speed. Further performance improvements come from a two stage algorithmic approach: dynamic programming and refinement. The algorithm is an order of magnitude faster than conventional approaches and correctly assigns 7-37% more amino acid residues in de novo sequences3.
Database search algorithms for protein identification.
The novor.cloud database search algorithm uses unique technology to improve the speed and accuracy of peptide identifications, even from the largest databases. De novo sequencing results are used to guide the database search, increasing accuracy and speed. Novor.cloud is 3X to 100X faster than other database search algorithms, and identifies 5% to 35% more acceptable peptides. Novor.cloud is the only search engine capable of searching the entire NCBI nr database of all known protein sequences.

Automatic parameter selection.
Novor.cloud contains technology to automate the search submission process
Post-translation
Whereas other de novo sequencing and protein ID software may struggle with or limit the number of post-translational modifications considered, novor.cloud allows you to turn on all possible PTM (even custom ones). Even after activating 41 PTMs, novor.cloud can complete the search 43% faster than another search engine4.
Error tolerance
Eliminates fine-tuning error tolerance parameters to balance sensitivity vs. speed and accuracy. Based on first-pass data analysis, examines the mass error present and adjusts further analysis accordingly. With no reasonable guidance given on search parameters, 90% of the search results are identical to results generated using ‘expert’ human guidance4.
Enzyme detection
Experimental conditions such as the enzyme used for digestion can be automatically detected in a first-pass analysis, prior to deeper analysis5.
Citations.
- Ma, Bin. “Novor: real-time peptide de novo sequencing software.” Journal of the American Society for Mass Spectrometry 26.11 (2015): 1885-1894
- Ma, Bin, et al. “PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry.” Rapid communications in mass spectrometry 17.20 (2003): 2337-2342.
- Zhang, Jing, et al. “PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification.” Molecular & cellular proteomics 11.4 (2012).
- Reinhardt et al. Cloud-based Software for Rapid and Accurate Peptide Identification and De Novo Sequencing, 2021 ASMS Poster, Abstract ID 30823
- Gholamizoj et al. Automatic Detection of the Protease used in Bottom-Up Proteomics Experiments, 2022 ASMS Poster, Abstract ID 309382